Prions are believed to be the cause of an array of rare but horrifying neurological diseases, such as Variant Creutzfeldt-Jakob disease (known, in cattle, as mad cow disease).
These misfolded proteins essentially eat microscopic moth holes into the brain. They are untreatable and always fatal.
Researchers at Imperial College London and the University of Zurich have now identified a critical step in the misfolding that creates a prion.
They were also able to halt the process, in a Petri dish, using antibodies — paving the way to possible treatments.
“Prion diseases are aggressive and devastating, and currently there is no cure,” Imperial’s Alfonso De Simone, the study’s lead researcher, said in a release.
“Discovering the mechanism by which prions become pathogenic is a crucial step in one day tackling these diseases, as it allows us to search for new drugs. Now we know what we’re targeting, we know what features drugs need to have to stop prions becoming pathogenic.”
A Nasty Twist: Proteins are the essential unit of life — complex molecules folded up with more turns, curls, and hairpins than a Hot Wheels track. Those folds determine the proteins’ jobs and functions, which are … well, everything. Proteins are the physical stuff that makes up tissues, cells, signaling chemicals, enzymes, you name it.
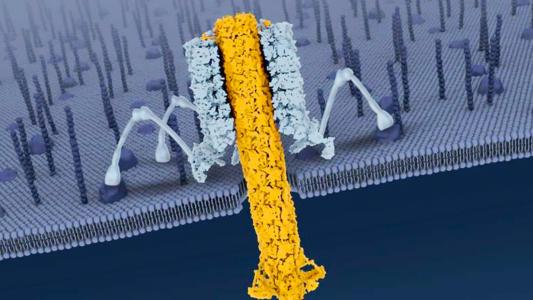
Prions are proteins gone rogue, twisted like a comic book villain into pathogenic shapes. Even worse, a prion can twist other proteins it meets into its misshapen image.
As single proteins, prions are unimaginably tiny — tinier than cells, bacteria, and even viruses — but they are capable of causing devastating diseases. Usually, prion diseases occur spontaneously or from inherited genetic mutations, but rarely, they can be transmitted through contaminated food, blood, or surgical instruments.
These are called transmissible spongiform encephalopathies, the most famous of which is bovine spongiform encephalopathy — aka mad cow.
That “spongiform” part is a funny word for a very unfunny condition. The prions cause a cascade of misfolding that basically turns your brain into a sponge, riddled with tiny holes.
Luckily, human prion diseases are quite rare; Johns Hopkins pegs the number of cases at around 300 a year in the U.S. These diseases include kuru — originally identified among populations that practiced ritual cannibalism, and now all but eliminated — and the most common form in humans, Creutzfeldt-Jakob disease (CJD).
Human prion protein naturally occurs in the body, but it is “usually well behaved,” says Valerie Sim, an associate professor at the University of Alberta’s Centre for Prions and Protein Folding Disease, who is not affiliated with the Imperial College/Zurich study.
But sometimes (hence the name), prion protein will twist the wrong way, triggering a domino effect that ends in disease and death.
It’s that moment, when the prion breaks bad, that the researchers were looking to figure out.
Into the Fold: For their study, published in PNAS, the researchers compared normal human prion protein with a mutated, pathogenic version.
The mutant prion protein was chosen for its aggressive, contagious nature — more prions equals more opportunities to try and catch them in the act. To do so, the researchers used advanced imaging techniques backed up with computer analysis.
The researchers found a specific spot on the prion where it began to fold into the pathogenic form. The University of Zurich team then produced antibodies that took aim at that exact spot. When the mutant prions were exposed to the antibodies in a test tube, they did not fold into their pathogenic shape.
“That supported their hypothesis that this spot on the mutant protein is the driver of misfolding,” Sim says.
The antibody’s success also serves as a proof-of-concept, of sorts, for potential prion therapies that take aim at folding sites.
“Now (researchers) are saying ‘we think it’s this specific spot on the prion protein that’s the trouble spot,'” Sim says. If a molecule could be found that blocks or stabilizes that spot, it could finally provide a prion treatment.
“How you do that is a whole other level of investigation,” Sim says.
Besides being performed in vitro, the study comes with a few other caveats. While the site the researchers identified may be the starting point for the mutated prion protein’s bad fold, it’s not necessarily the same place it happens in the regular human prion protein without the same mutation.
Also, while the team’s mutated prion protein is known to cause disease in humans, the researchers did not infect an animal model with their proteins, so they didn’t prove the ability of these specific prions to cause disease. Unlike a virus, this can’t be determined just by looking at it: it’s actually pretty tricky business growing a pathogenic prion, Sim says — usually, you need to use slurried infected brain matter.
Still, the finding may prove an important insight into the unusual pathogens.
“The intermediate stage of prion pathogenesis is so transient it’s like a ghost — almost impossible to image,” study lead author Máximo Sanz-Hernández says in the release.
“But now we have a picture of what we’re dealing with, we can design more specific interventions that can one day potentially control these devastating diseases.”
We’d love to hear from you! If you have a comment about this article or if you have a tip for a future Freethink story, please email us at [email protected].